"HIPAA, FDA SaMD, EU AI Act, state licensure, CHAI. Five regulatory layers, one LLM deployment, and a permanent need for caution."
Compass, Regulatory-Stack-Climber AI Agent
Healthcare LLM deployment in the U.S. is governed by an overlapping framework of FDA medical-device rules, HIPAA privacy obligations, state licensure laws, malpractice-insurance requirements, and emerging multi-stakeholder assurance standards. Internationally the picture is similar in shape but different in detail: EU AI Act high-risk classification, MDR/IVDR for devices, GDPR special-category provisions for health data, NHS data-protection rules in the U.K. Every production deployment touches at least three of these frameworks; any deployment that is patient-facing touches all of them. This section maps each track to the deployment pattern that satisfies it.
Prerequisites
This section assumes the healthcare-LLM failure modes from Section 69.2, the LLM-policy vocabulary from Section 55.1, and the audit-log discipline from Section 54.9.
FDA Software as a Medical Device (SaMD)
The FDA's PCCP (predetermined-change-control-plan) framework was finalized in January 2025 after roughly seven years of drafting. The first FDA-approved SaMD with a PCCP was an AI-based diabetic-retinopathy screening tool called IDx-DR (now LumineticsCore), which was originally cleared in 2018 and spent six years waiting for a regulatory framework that would let it update its model without re-clearance.
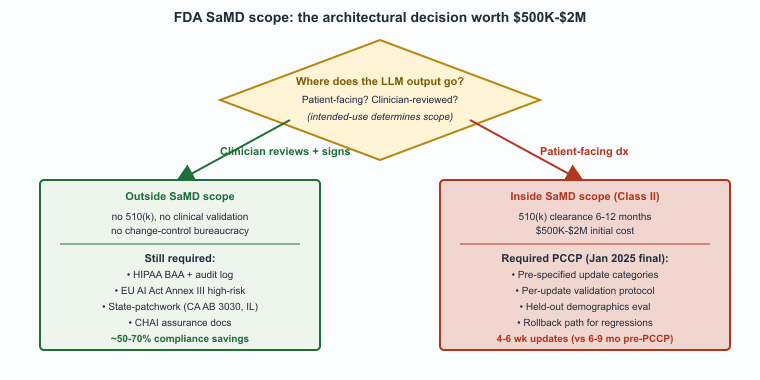
Whether a clinical LLM is a regulated medical device depends on intended use. "Provides clinical decisions a clinician must independently review" is generally not a device under the FDA's SaMD framework; "provides patient-facing diagnoses" is. The 2024-2026 FDA guidance on AI/ML SaMD has clarified the predetermined-change-control-plan (PCCP) framework, allowing post-market model updates without re-certification, which is a major practical improvement for LLM products that improve frequently. The FDA's January 2025 final guidance on PCCPs is the document to read; the framework allows vendors to pre-specify the kinds of model updates they will make and how they will validate them, which removes the previous bottleneck of full re-clearance for every model improvement.
HIPAA and Equivalent Privacy Regulations
HIPAA (U.S.) and equivalent privacy regulations require Business Associate Agreements with all LLM providers, audit logs, and the minimum-necessary rule on retrieval indexes. The practical implication for procurement: any LLM provider whose service touches protected health information needs a signed BAA, the BAA must cover the specific service (not just the cloud platform), and the BAA must specify retention, training, and de-identification posture. Major LLM providers (Azure OpenAI Service for Healthcare, AWS HealthLake, Google Cloud Healthcare API) offer healthcare-specific SKUs with HIPAA-eligible terms; using anything weaker for clinical data is a compliance defect.
EU AI Act High-Risk Classification
Most clinical-use LLMs are high-risk under the EU AI Act Annex III, point 5 (access to essential services, including healthcare). Conformity assessment, post-market monitoring, and human oversight requirements apply. For products that are also medical devices under MDR/IVDR, the AI Act overlays an additional regime. The practical impact is meaningful documentation overhead: technical documentation, risk-management files, post-market surveillance plans, and a registered EU representative for non-EU vendors.
State-Level Licensure Rules
Several U.S. states have introduced legislation specifically targeting AI in clinical practice; the patchwork is evolving rapidly. California's AB 3030 (2024) imposes disclosure obligations on AI-generated patient communications. Texas's SB 1188 (2023) addresses AI use in mental-health contexts. Illinois has proposed specific rules around AI-generated clinical decision support. The pattern: state laws often impose more specific obligations than federal frameworks, particularly around disclosure and informed consent, and multi-state health systems must support per-jurisdiction configuration.
Malpractice Insurance
Major medical-malpractice insurers have begun pricing AI usage into premiums; some require disclosure of AI tools in standard practice. The pricing impact varies by use case: ambient documentation is broadly seen as risk-neutral or risk-reducing (the documentation quality is better), while patient-facing autonomous chatbots are priced as higher-risk. The insurer expectations have started to look like compliance requirements in their own right: an insured practice using an LLM tool without an audit log and a documented human-review workflow risks coverage denial in a tail event.
Multi-Stakeholder Consensus Standards
The Coalition for Health AI (CHAI) publishes assurance-standards drafts that increasingly serve as the de-facto baseline for U.S. hospital procurement. CHAI's framework covers model cards, bias evaluation, post-market monitoring, and explainability; CHAI-aligned RFPs are now common at major U.S. academic medical centers. The standards are not legally binding but have become a procurement requirement; vendors that cannot produce CHAI-aligned documentation are eliminated early in major-system selection. The eHealth Initiative publishes a parallel framework focused on operational deployment. The NIH and ONC have signaled support for the multi-stakeholder approach.
The interaction between FDA SaMD scope and the human-in-the-loop posture is the load-bearing engineering decision in U.S. healthcare LLM design. A product designed as "assistive, clinician makes the decision" generally stays outside SaMD scope; a product designed as "produces patient-facing recommendations" generally enters it. The cost of being inside SaMD is substantial (regulatory affairs team, clinical validation, post-market surveillance, change-control bureaucracy); the benefit is that the product can do things that a non-device cannot (give direct patient guidance, replace clinician judgment in narrow scopes). Most LLM vendors in 2026 design intentionally to stay outside SaMD by keeping the clinician in the loop, which both reduces regulatory burden and matches the clinical-practice norms that practicing physicians actually want.
International Variation
Outside the U.S. and EU the picture varies. The U.K.'s MHRA has issued specific guidance on AI as a medical device, broadly aligned with FDA's framework. Canada's Health Canada follows MDR-style rules. Singapore's HSA has been an early adopter of risk-tiered AI guidance. China's NMPA has issued AI medical-device guidance distinct from Western frameworks. Multi-jurisdiction deployments must navigate each; most vendors release country-by-country rather than attempting global launches.
Who. A mid-stage clinical-AI vendor offering an LLM-augmented radiology-report-drafting tool that interprets free-text findings against structured clinical reasoning. Situation. The product is classified as Class II SaMD by the FDA because its outputs can materially influence the radiologist's report and downstream care decisions. Problem. Pre-2024, every model update (improved base model, retraining on additional data, hyperparameter changes) required a 510(k) supplemental submission and 6-9 months of FDA review, leaving the product locked to base-model versions 12-18 months behind the frontier. Decision. The vendor filed a Predetermined Change Control Plan (PCCP) under the FDA's January 2025 final guidance, specifying in advance the categories of changes they would make (base-model upgrades within an architectural family, retraining on additional de-identified data from existing institutions, prompt updates within bounded scope) and the validation protocol for each. How. The PCCP defines fixed performance thresholds (sensitivity, specificity, demographic-stratified accuracy), a held-out evaluation set that triggers automatic re-validation, and a documented rollback path for regressions. Result. Post-PCCP, model updates ship in 4-6 weeks rather than 6-9 months. Lesson. The PCCP framework converts the regulatory bottleneck from per-update review to per-update validation, which is the difference between annual and quarterly capability refresh in regulated clinical AI.
Stacking the regulatory frameworks produces concrete numbers. FDA SaMD 510(k): typical clearance time 6-12 months, total compliance cost (clinical validation, regulatory affairs, technical documentation) of $500K-$2M for a Class II product, dropping by roughly 40-60 percent on subsequent submissions inside a PCCP. HIPAA breach exposure: per-violation-type annual cap of $1.99M, average healthcare breach cost of $10.93M (IBM 2024). EU AI Act high-risk: conformity assessment plus post-market monitoring adds roughly 4-8 months of pre-market time and 0.5-1.5 FTE/year of ongoing compliance staff for a small vendor. State patchwork: California AB 3030 disclosure and Illinois mental-health-AI rules together add roughly 0.25 FTE/year of legal and engineering effort to maintain per-jurisdiction configurations.
Aggregated, a clinical-AI vendor selling into the U.S. and EU should budget $1-3M in initial compliance spend, 2-3 FTE of ongoing compliance and regulatory affairs at a $200K fully-loaded rate ($400-600K/year), and 6-12 months of pre-market time beyond the engineering build. The cost is consequential but predictable; the architectural decisions that keep the product outside SaMD scope and inside BAA-covered cloud (Section 69.1) typically reduce these numbers by 50-70 percent, which is why "stay outside SaMD" is the dominant design strategy.
- Chapter 53 (Regulation and Compliance) for the cross-cutting regulatory framing across LLM verticals.
- Section 53.4 (Licensing, IP, and Privacy) for the BAA and licensing-framework details that operationalize HIPAA contractual posture.
- Chapter 42 (Evaluation Foundations) for the held-out evaluation methodology that PCCP frameworks depend on.
- Section 67.2 (Legal Regulatory Framework) for the parallel regulatory structure in the legal vertical.
- Section 68.3 (Finance Regulatory Framework) for the model-risk-management parallel from finance.
Show Answer
Show Answer
Show Answer
What Comes Next
Section 69.4 covers the four HIPAA-compliant deployment patterns that have consolidated for U.S. healthcare: BAA-covered cloud, de-identified-then-cloud, VPC-isolated cloud, and on-premises open-weight. The choice among them is the dominant architectural decision in a U.S. healthcare LLM project.
What's Next?
In the next section, Section 69.4: HIPAA-Compliant Deployment Patterns, we build on the material covered here.